| A 53 year-old woman developed a rapidly progressive dementia over three months associated with prominent myoclonic jerks. Over the following month, she became bed bound and mute. |
![]()
![]()
![]()
![]()
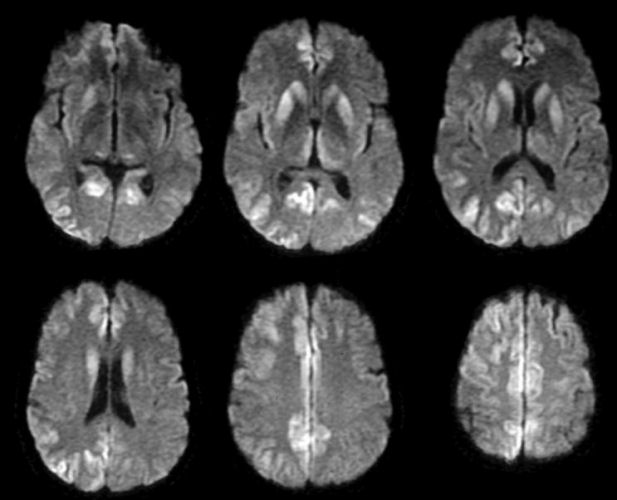
| Creutzfeldt-Jakob Disease (CJD): Axial diffusion-weighted
MRIs. Note the numerous areas of high intensity in the caudate, putamen and medial thalamus bilaterally, as well as
the cortical lesions posteriorly and midline along the cingulate gyrus. These diffusion-weighted
images are consistent with the clinical diagnosis of CJD.
CJD is a transmissible, progressive, fatal spongiform encephalopathy. The cardinal features of the disease include a rapidly progressive dementia, generalized myoclonus, and a characteristic EEG pattern of periodic sharp wave complexes. Patients typically present with rapidly progressive dementia, cerebellar dysfunction, visual disturbances, or a combination of all three. The infectious agent is a prion (protein) that can be transmitted either by direct contact with contaminated tissue (iatrogenic) or via inheriting a mutation in the prion protein gene (familial). However, most cases of CJD are sporadic. Prion disorders in animals include scrapie in sheep and bovine spongiform encephalopathy in cattle (aka BSE or "mad cow" disease). In humans, familial forms include familial CJD, fatal familial insomnia, and Gerstmann-Straussler-Scheinker Syndrome. Another human form, Kuru, resulted from cannibalistic practices in New Guinea. Ingestion of tainted beef has been linked to so-called Variant Creutzfeldt-Jakob disease (vCJD). Formerly, brain biopsy was required for diagnosis. Now, MRI scans, especially Flair and diffusion-weighted images can be used. If they show the characteristic picture above, in the correct clinical context, the diagnosis becomes more straightforward. |
Revised
11/30/06
Copyrighted 2006. David C Preston