I am currently a doctoral trainee in Dr. Ruth Keri's lab. The primary goal of the Keri laboratory is to discover molecular mechanisms that drive breast cancer initiation and progression with the ultimate intent of identifying novel therapeutic approaches to treat this diverse collection of malignancies. A major area of focus involves discerning the molecular underpinnings of chromosomal instability and how this aspect of breast cancer can be therapeutically leveraged to improve patient outcomes.
Chromosomal instability (CIN) is a characteristic of many cancers that contributes to a tumor cell’s accumulation of genetic defects including mutations, copy number alterations, and aneuploidy. At low levels CIN can promote tumor evolution by conferring a selective advantage to tumor cells growing in diverse microenvironments and in the presence of various therapies. Triple Negative Breast Cancers (TNBCs) typically have high rates of CIN, and this facilitates disease aggressiveness and poor patient outcomes. However, cells must strike a balance between evolution and homeostasis. While basal levels of CIN contribute to cancer progression, excessive CIN leads to cell death due to the loss or mutation of essential viability genes. Discovering proteins necessary for cells to maintain basal, tumor-promoting levels of CIN that can be leveraged to induce catastrophic CIN and cell death should reveal therapeutic vulnerabilities in breast and other cancers for which CIN is a hallmark phenotype.
We have identified Structural Maintenance of Chromosomes 2 (SMC2) as a modulator of CIN in TNBC. SMC2 is a core member of condensin, a protein complex that is largely known for its role in maintaining chromosome architecture, particularly during mitosis. SMC2 has also recently been implicated in the regulation of transcription, suggesting that this protein may lie at the interface of transcription and chromatin configuration to regulate CIN. Our studies have revealed that SMC2 is required for long term TNBC cell viability and proper progression through the cell cycle. Further, SMC2 silencing results in an increase in CIN phenotypes including DNA damage, multinucleation, dysmorphic nucleation, and chromatin bridges. Over time, these defects accumulate to an excessive level that ultimately induces TNBC cell death. Notably, SMC2 is overexpressed in a subset of TNBC patient tumors. SMC2 overexpression is associated with an increased number of mutations, copy number alterations, and aneuploidy, as well as worse patient outcomes, implicating SMC2 as a therapeutically relevant target. Together, these results indicate that SMC2 regulates CIN in TNBC. We conclude that SMC2 is a potential target for therapeutic development for the treatment of TNBC, an aggressive disease with limited therapeutic options.
| Control Cell | Chromosome Bridge |
|---|---|
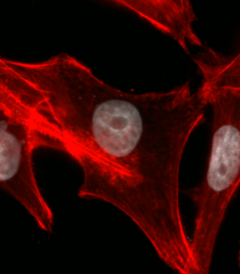
|
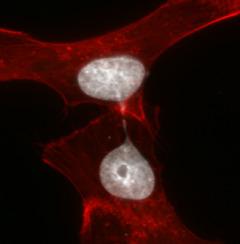
|
| TNBC cells were transfected with non-silencing siRNA (siNS) or siRNA targeting SMC2 (siSMC2) for up to 4 days. Cell bodies were stained with Phalloidin (red) and nuclei/DNA were stained with DAPI (white). Representative images of a control cell with a normal nucleus (left) and SMC2 silenced cells with a chromatin bridge (right). | |