Using cryo-electron microscopy, researchers at Case Western Reserve University School of Medicine and National Institutes of Health open door to new level of detail in study of abnormal proteins that cause untreatable neurodegenerative conditions
The highest-ever resolution imaging of an infectious prion has provided the first atomic-level data of how these abnormal proteins are assembled to cause fatal neurodegenerative diseases in people and animals—and how they can be potentially targeted by new therapies.
Conducted by Case Western Reserve University and the National Institutes of Health (NIH), the research is available through Molecular Cell. (The DOI for the paper: 10.1016/j.molcel.2021.08.011.)

“These detailed prion structures provide a new premise for understanding and targeting these currently untreatable diseases,” said Allison Kraus, lead and co-corresponding author of the research and an assistant professor in the Department of Pathology at Case Western Reserve University School of Medicine. “It will now be much easier to develop and test hypotheses about how prions are assembled as highly infectious and deadly protein structures.”
Seeing the basic building blocks of these lethal proteins, she said, provides a foundation for therapeutic strategies to block the spread, buildup and toxicity of prions.
Prions are proteins in brain tissue that transmit their irregular “misfolded” shapes onto the regular version of the same protein—and are the source of mammalian diseases, including human conditions like Creutzfeldt–Jakob disease (CJD) and its variant, known as vCJD, as well as Gerstmann–Sträussler–Scheinker syndrome, and others.
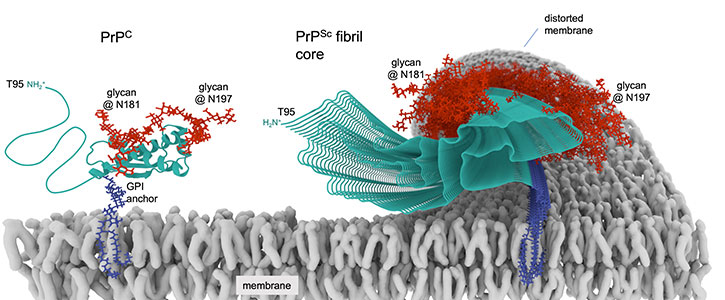
shown tethered to a cell membrane beside the
corrupted infectious form that forms a prion fibril.
Similar prion-like mechanisms occur in the characteristic proteins suspected in the development of other neurodegenerative conditions, including Parkinson’s disease, Lou Gehrig’s disease (also known as ALS, or amyotrophic lateral sclerosis), chronic traumatic encephalopathy (CTE) and Alzheimer’s disease.
Though instances are rare, prion diseases can be transmitted between people; others are readily transmissible between animals, such as chronic wasting disease.
For this study, researchers imaged rodent-adapted scrapie prions derived from the brains of clinically ill hamsters.